KisSplice2RefTranscriptome (K2rt)
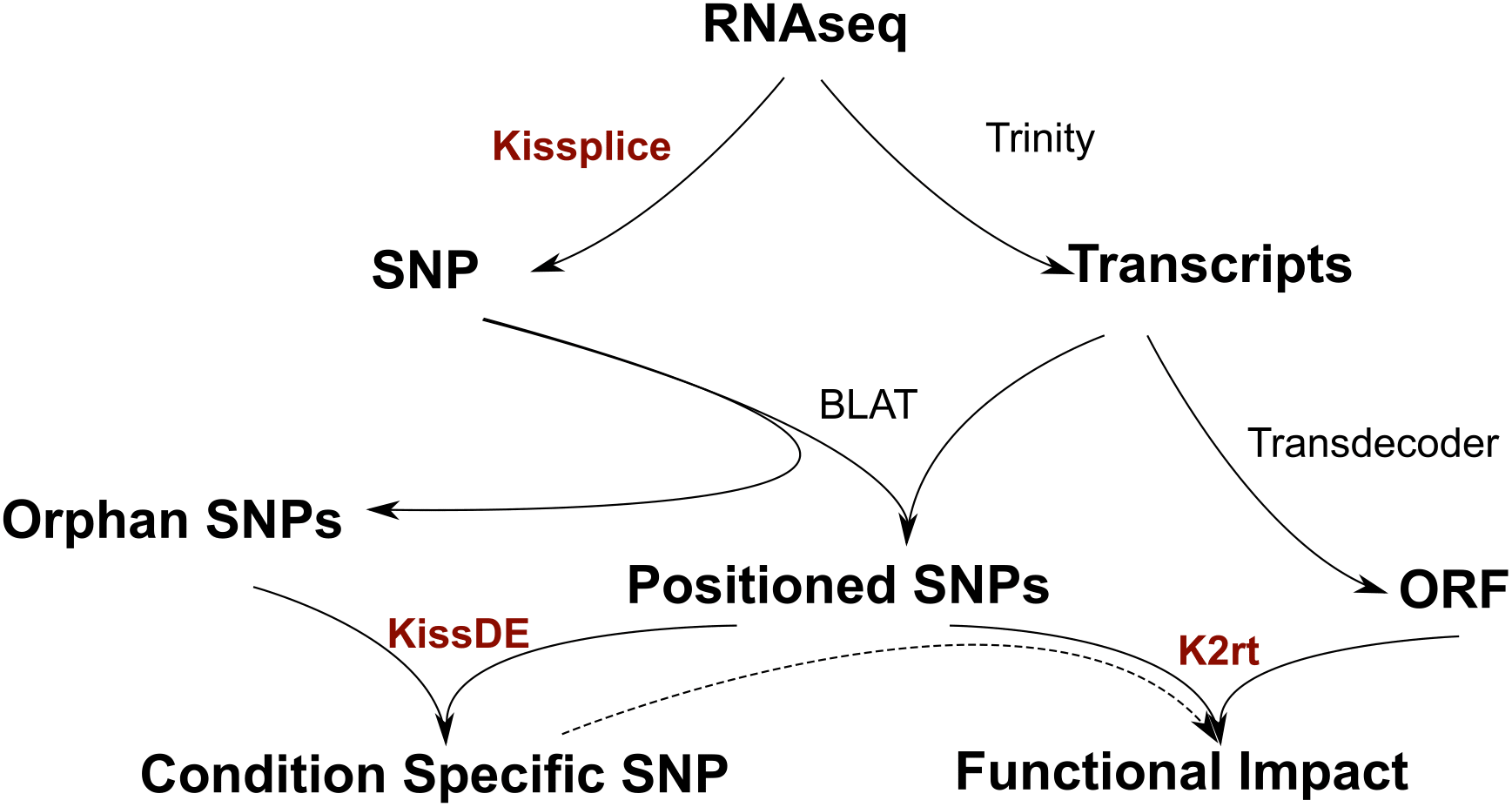
With fasta/fastq input from an RNA-seq experiment, SNPs are found by KisSplice without using a reference. KisSplice provides only a local context around the SNP, but a reference transcriptome can be built from the RNAseq data using a full-lenth transcriptome assembler like Trinity. Then SNPs predicted by KisSplice can be positionned along the transcripts (with BLAT). Some SNPs that do not map on the transcripts of Trinity, called orphan SNPs, are harder to study but can still be of interest. We propose a method, KisSplice2RefTranscriptome, to predict a functional impact for the positioned SNPs, and intersect these results with condition-specific SNPs. Overall, starting from RNAseq data only, we obtain a list of condition-specific SNPs stratified by functional impact.
Version 1.3.3
KisSplice2RefTranscriptome version 1.3.3 (2019-07-20)User's guide
- Bug fixed
- When using >30 conditions with KisSplice, the fasta header is very long, and blat truncates it when producing .psl files, which causes downstream issues in k2rt. This bug was initially reported here https://www.biostars.org/p/384191/
Older versions :
Version 1.3.2
KisSplice2RefTranscriptome version 1.3.2 (2017-10-24)
- Improvements
- The -pval option was added (optional ; KissDE p-value cutoff - default : 0.05)
- K2RT output now has two more columns : KissDE_p-value and KissDE_DeltaF, they contain respectively t$
Version 1.3.1
KisSplice2RefTranscriptome version 1.3.1 (2017-07-18)
- Improvements / Bugs fixed
- We now perform a test to check that the value given for parameter -K and the k-mer size used to run KisSplice are identical.
Version 1.3.0
KisSplice2RefTranscriptome version 1.3.0-p1 (2017-04-02)
Version 1.2.2
KisSplice2RefTranscriptome version 1.2.2 (2016-04-22)
Requirements
KisSplice2RefTranscritpome is written in Python and is running on Linux platforms. A version of python 2.7 or more is needed.
How to get my input files ?
Running KisSplice
- The -pval option was added (optional ; KissDE p-value cutoff - default : 0.05)
- K2RT output now has two more columns : KissDE_p-value and KissDE_DeltaF, they contain respectively t$
- We now perform a test to check that the value given for parameter -K and the k-mer size used to run KisSplice are identical.
kissplice -s 1 -k 41 -r sample_example/reads1.fq -r sample_example/reads2.fq -r sample_example/readsN.fq
Trinity ORFs file
Here is a basic example command in order to run Trinity :trinity --seqType fq --left reads_lefts.fq --right reads_right.fq
The assembled transcripts will be written in the Trinity.fasta file. You will then need to run Transdecoder (included in the Trinity package). Transdecoder will predict the ORFs of the transcripts that will be used for the prediction of the functional impact of SNPs. Here is a basic command in order to run Trandecoder on Trinity ouput :Trandecoder.LongOrfs -t Trinity.fasta
Trandecoder will output several files, KisSplice2RefTranscritpome only needs the .bed file.Running BLAT
KisSplice2RefTranscriptome needs the bubbles to be postioned on the reference transcriptome. For now, only psl formated files are supported by K2rt. You can run BLAT to get this file. Here is a command in order to run BLAT :blat --minIdentity=80 Trinity.fasta kissplice_output_type0_coherent.fa output.psl
Running KissDE (optional)
You can intersect KisSplice2RefTranscriptomeresults with condition-specific SNPs by giving to K2rt the ouput of kissDE.Using KisSplice2RefTranscriptome
You have access to all options description with the help command :
Usage: kissplice2reftranscriptome [-h]
-b PATH_TO_TRINITY_FILE
-k PATH_TO_KISSPLICE_FILE
-t PATH_TO_BLAT_FILE
[-s PATH_TO_KissDE_FILE]
[-o PATH_TO_MAIN_OUTPUT_FILE]
[-l PATH_TO_LOW_QUERY_COVERAGE_OUTPUT_FILE]
[-m PATH_TO_MERGED_BUBBLES_OUTPUT_FILE]
[-u PATH_TO_UNTREATED_BUBBLES_OUTPUT_FILE]
[-Q QC_MIN] [-C KC_MIN] [-K K_VAL]
[--merge_codon]