Frequently asked questions
- What is a bubble and what is the difference with a BCC?
- Can sequencing errors create bubbles?
- How can I differentiate a SNP from a sequencing error ?
- How can I differentiate a SNP from an inexact repeat ?
- How can I differentiate a SNP from RNA editing?
- How can I differentiate an AS from an indel ?
- Why are there incoherent bubbles ? Aren't all bubbles created by the reads ?
- What are the "type 4" events ?
- What are the counts given by KisSplice?
- Can paralog genes create bubbles?
- Why are there duplicated events in the output?
- I have several experimental conditions and I want to find SNPs whose allele frequency change significantly accross conditions.
- In KisSplice2refTranscriptome output (version < 1.3.1), what is the meaning of the columns
bubble_aligned_in_multiple_comp and bubble_aligned_in_multiple_seq ? - When I change the value of k, the number of SNPs changes a lot. Which value should I keep ?
- How to visualise my SNPs ?
KISSPLICE: de-novo calling alternative splicing events from RNA-seq data
SNP calling from RNA-seq data without a reference genome
Question not in the list? Ask your own!
KISSPLICE: de-novo calling alternative splicing events from RNA-seq data
-
What is a bubble and what is the difference with a BCC?
- What is a bubble ?
A bubble corresponds to a variation in the sequence. Formally, it is pair of disjoint paths with the same source and target nodes in the de Bruijn graph (DBG) derived from the reads. Depending on the length of the paths of the bubble, the variation will be a SNP, an indel or an AS event. Repeats may also generate bubbles, but in this case, the paths will not have the same length and will have high sequence similarity.

Here, an example of an alternative splicing event or an indel. These paths have the same sequences on the borders (a and b) but vary in the center (junction ab for the shorter path, junction aS, exon S and junction Sb for the longer path).
KisSplice will output the two paths of the bubble (here ab and aSb). Every output is pairwise. There will be the sequences in a fasta file, with the identifiers:
bcc_X|cycle_Y|type_Z|upper_path_length_L ...
bcc_X|cycle_Y|type_Z|lower_path_length_L ...
X is the bcc identifier, Y is the bubble identifier, Z is the type of event detected, L is the length of the sequence. Z can be 0 for SNPs and sequencing errors, 1 for alternative splicing events, 2 for inexact tandem repeats, 3 for short indels. - What is a BCC ?
A BCC corresponds to a set of overlapping variations. In graph theory, a BCC is a BiConnected Component (maximal subgraph such that there are two paths between any two nodes). In KisSplice, a BCC is therefore a biconnected component of the de Bruijn graph derived from the reads. Each BCC contains at least one variation (SNP, indel, AS event, etc.). It may contain more than one variation if they overlap, for instance when a SNP is embedded in a skipped exon.
When a gene contains several variations, but distant by more than k nt, it will be split among several BCCs. Hence, a BCC does not correspond to a gene, but to a gene fragment.
If two genes share k nt, they will fall in the same connected component, not necessarily the same biconnected component. If they share at least 2 k-mers, then the region inbetween will form a BCC whose gene affiliation is unclear. The genes are said to be co-assembled. This is the case for instance for (recent) paralogous genes.
- What is a bubble ?
-
Can sequencing errors create bubbles?
Yes, they can. If two reads differ by one nucleotide, it could create a bubble and be classified as type 0. In practice, at least two reads are required (-c option) to call a bubble.
-
How can I differentiate a SNP from a sequencing error ?
In KisSplice, there are two filters which are meant to filter out sequencing errors. The first is an absolute filter (-c option) and removes each kmer seen less than c times. The second is relative (-C option) and removes, for each kmer, every outgoing (resp incoming) edge covered by less than C% of the sum of coverage of its outgoing (resp. incoming) edges.
By default, c=2 and C=5%. If the goal is to identify unfrequent variants, these parameters can be lowered, but replicates are then recommended. The rationale is that the same sequencing error should not appear at the same location is multiple replicates.
We choose not to use the quality because some PCR artifacts can generate high-quality sequencing error.
-
How can I differentiate a SNP from an inexact repeat ?
This is very difficult, especially if the copies of the repeats diverged recently.
An inexact repeat consisting of two identical regions of 2k+1 nt which diverged only in their central position will be strictly identical to a SNP in the DBG.
In practice, many inexact repeats correspond to larger divergence levels (divergent sites are located less than 41nt apart) and therefore will lead type0b bubbles, or no bubble at all (if there the two copies of the repeat do not share at least 2 kmers). Filtering out type0b bubbles is a simple and efficient strategy to get rid of most inexact repeats... at the expense of missing true SNPs located in a cluster.
Another useful idea is to rely on the differential analysis. If you have several biological conditions, and if one variant is enriched in one condition and the other variant is enriched in the other condition, this is an indication that either it is a real SNP, or it is an inexact repeat where one copy is condition-specific (which itself is also interesting).
-
How can I differentiate a SNP from RNA editing?
You cannot if you have only RNA-seq data.
-
How can I differentiate an AS from an indel ?
If there is a reference genome, it is possible to re-align the paths and check the real nature of the event. By default, KisSplice does a classification based on the length of the variable part. If the variable part has length 1, 2, 4, 5, then the event is classified as an indel. Otherwise, it is classified as an alternative splicing event.
The motivation for this choice is based on the two following observations :- indels tend to be short
- AS events tend to fall in CDS, not UTRs, hence tend to have a variable region whose size is a multiple of 3
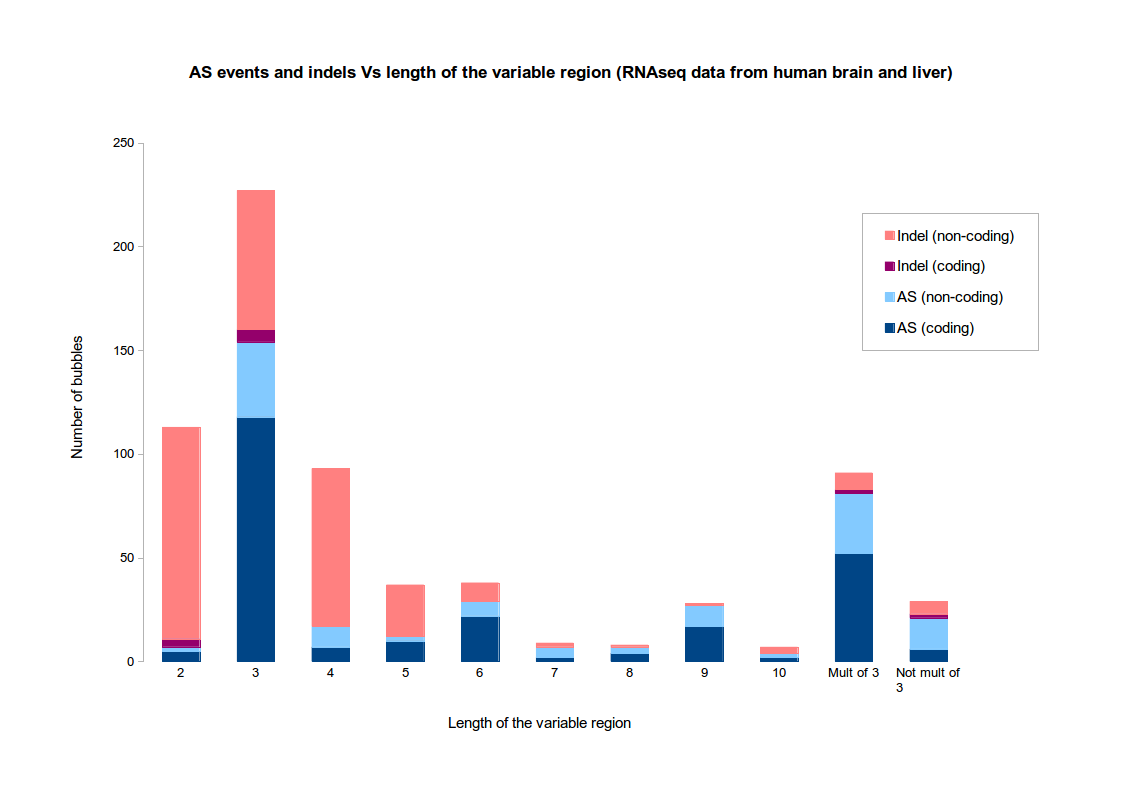
The figure illustrates these observations on an RNAseq dataset from human tissues (brain and liver). This dataset is the one used in the original KisSplice paper. Clearly, when the variable part has size exactly 3, it is difficult to assess if the variation is an AS event or an indel.
-
Why are there incoherent bubbles ? Aren't all bubbles created by the reads?
A bubble is incoherent if there are no tilings of reads covering each path of the bubble. Most incoherent bubbles are artifacts of the DBG. They are due to the fact that DBG lose the information that two k-mers are in the same read. In practice, some incoherent bubbles can correspond to true variations in poorly expressed regions.
-
What are the "type 4" events ?
They are bubbles which can not be classified as type 1, 2, or 3 because they do not satisfy the criteria. For instance, they may correspond to cases where a SNP is close to the alternative splice site, hence generating a path of length larger than 2k-2.
What are the counts given by KisSplice?
The counts correspond to the number of reads that align to each path. If a read aligns to the border of a path with less than k nt, it is discarded.
-
Can paralog genes create bubbles ?
Yes they can, if they did not diverge too much. In this case, they fall in the category of inexact repeats, which indeed may generate bubbles which resemble SNPs. In this case, focusing on condition specific bubbles (one member of the gene family enriched in one dataset, and the other enriched in the other dataset) is still relevant.
It is worth outlining that in the case of paralog genes, it is often the case that the copies differ by more than one position. If these positions are distant by more than k nt, this will generate several bubbles. Otherwise, this may generate a single bubble, but whose paths will be longer. In the case where the copies do not share at least two k-mers (i.e. the copies diverged too much), then no bubble is reported.
In practice, long bubbles of type 0 are often good indicators of paralogs. Of course, they may also correspond to clusters of true SNPs. Again, focusing on condition specific bubbles is one way to focus on the most interesting bubbles.
In the case where at least one member of a gene family is alternatively spliced, it will generate a bubble. If the copies of the gene family did not diverge much, it may be that it is impossible to decide which member of the gene family contibuted to which alternative transcript. In this case, the gene family can be thought of as "collectively spliced". Why are there duplicated events in the output?
Looking at KisSplice outputs, some events may appear as duplicates. It may very well be that they differ only by one nt. This happens when a SNP/mutation is coupled with other events (such as AS events).
For example, in the case of an intron retention, the intron may accumulate many mutations. This will create many bubbles which will all be reported by KisSplice.
SNP calling from RNA-seq data without a reference genome
See also our K2rt page the user's guideI have several experimental conditions and I want to find SNPs whose allele frequency change significantly accross conditions.
You should use KissDE, and select in the output SNPs whose p-value is below a threshold. You can then rank the SNPs by ascending DeltaF (Difference of allele frequency accross conditions). You can focus on the first ones. The p-value and DeltaF columns can be found both in the output of KissDE and K2rt. Example of how to run KissDE and K2rt are given here and here.
In KisSplice2refTranscriptome output (version < 1.3.1), what is the meaning of the columns bubble_aligned_in_multiple_comp and bubble_aligned_in_multiple_seq ?
Indeed the names were not very explicit. We now renamed them SNP_in_multiple_assembled_genes and SNP_in_multiple_assembled_isoforms. SNPs mapping to several genes are likely to be false positives due to inexact repeats (for instance hidden paralogy). Those can be filtered out. SNPs mapping to several isoforms of the same gene are reported several times in the output of k2rt. The impact on the protein may be different from one isoform to another. If you are not interested in this level of detail, you can keep only one line for each SNP.
The following code performs these two filters :less mainOutput.tsv | awk '{multipleGenes=$11; multipleIsoforms=$12; SNPID=$2; if (multipleGenes=="False"){if (multipleIsoforms=="False"){print} else {if (alreadyprinted[SNPID]!=1){print; alreadyprinted[SNPID]=1}}}}' > mainOutput_no_multiple_genes_one_isoform_per_gene.tsv
We however would like to point out that Trinity, Oases, and other assemblers try to predict genes and isoforms, but they often fail. Recent paralogs tend to be assembled as multiple isoforms of the same assembled gene. On the other hand, a gene with a very high level of polymorphism, can be split in several assembled genes.
K2rt inherits from these errors in its classification. We advise users not to rely blindly on the notions of assembled genes and assembled isoforms.When I change the value of k, the number of SNPs changes a lot. Which value should I keep ?
The choice of k depends on the level of polymorphism of your species, and the precision/recall that you would like to obtain. The choice of k is a tradeoff between recall and precision. High values of k enable to predict few high confidence SNPs. Low values of k enable to predict more SNPs, at the expense of outputing false positives due to inexact repeats. In our paper (Lopez-Maestre et al. 2016, Supp Figure 3), we show that k=41 performs well in human (where polymorphism is low). For Asobara Tabida and Drosophila mojavensis/arizonae, k=41 gives a good precision, but the sensitivity is unknown. All SNPs located less than 41nt apart will be missed.
How to visualise my SNPs ?
We do not provide visual output for now. If you are interested in a specific assembled isoform/gene and you would like to produce a visualisation, we advise to map your reads to the assembled isoform and use IGV to visualise the .sam files. Coloured bases should correspond to your SNPs. In the case of a gene with several isoforms, you will need to visualise separately each isoform.