The pipeline
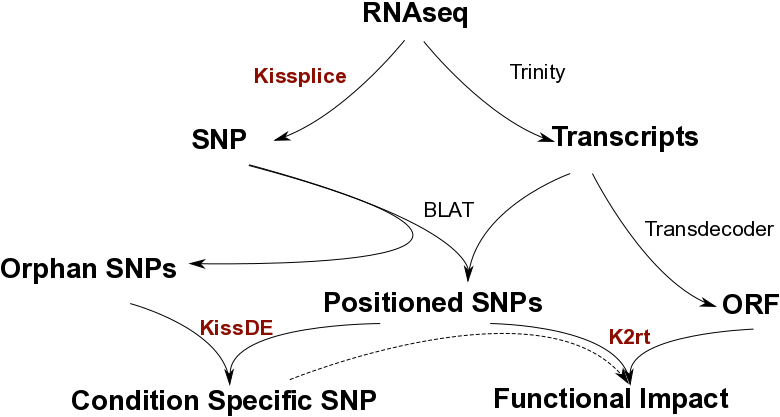
With fasta/fastq input from an RNA-seq experiment, SNPs are found by KisSplice without using a reference. KisSplice provides only a local context around the SNP, but a reference transcriptome can be built from the RNAseq data using a full-lenth transcriptome assembler like Trinity. Then SNPs predicted by KisSplice can be positionned along the transcripts (with BLAT). Some SNPs that do not map on the transcripts of Trinity, called orphan SNPs, are harder to study but can still be of interest. We propose a method, KisSplice2RefTranscriptome, to predict a functional impact for the positioned SNPs, and intersect these results with condition-specific SNPs. Overall, starting from RNAseq data only, we obtain a list of condition-specific SNPs stratified by functional impact.
KisSplice2RefTranscriptome
KisSplice2RefTranscriptome web pageKisSplice2RefTranscriptome version 1.2.0 (2016-29-03)
User's guide
Running the programs
Our method enables to find SNPs from RNA-seq data. In order to assess if the SNPs we find are correct, and if the list we output is exhaustive, we chose to test our method on RNA-seq data from the Geuvadis project. Indeed, the individuals whose transcriptome was sequenced in this project were already included in the 1000 genome project. Hence, their SNPs have been already well annotated. We downloaded fastq files from SRA and selected 10 Toscans and 10 Central Europeans. We sampled 10M reads for each individual and concatenated the fastq files in pools of 5 individuals.The files used in our analysis are available here
Running KisSplice
Here is the command in order to run KisSplice on this dataset :
kissplice -s 1 -k 41 --experimental
-r cond1_replicat1_R1.fastq -r cond1_replicat1_R2.fastq -r cond1_replicat2_R1.fastq -r cond1_replicat2_R2.fastq
-r cond2_replicat1_R1.fastq -r cond2_replicat1_R2.fastq -r cond2_replicat2_R1.fastq -r cond2_replicat2_R2.fastq
Trinity ORFs file
Here is the command in order to run Trinity on the human dataset:
trinity --seqType fq --JM 100G
--left cond1_replicat1_R1.fastq, cond1_replicat2_R1.fastq, cond2_replicat1_R1.fastq, cond2_replicat2_R1.fastq
--right cond1_replicat1_R2.fastq, cond1_replicat2_R2.fastq, cond2_replicat1_R2.fastq, cond2_replicat2_R2.fastq
--CPU 10 --bflyCPU 10
Transdecoder.LongOrfs -t Trinity.fasta
TransDecoder.Predict -t Trinity.fasta
Running BLAT
KisSplice2RefTranscriptome needs the bubbles to be positioned on the reference transcriptome. For now, only psl formated files are supported by K2rt. You can run BLAT to get this file. Here is the command in order to run BLAT on the human dataset :blat --minIdentity=80 Trinity.fasta results_cond1_cond2_k41_coherents_type_0.fa out_blat_SNP_on_trinity_ID_80.psl
The BLAT output can be downloaded here : out_blat_SNP_on_trinity_ID_80.pslRunning KissDE (optional)
You can intersect KisSplice2RefTranscriptome results with condition-specific SNPs by giving to K2rt the output of kissDE.
#!/usr/bin/Rscript
library(kissDE)
snp<-kissplice2counts("results_cond1_cond2_k41_coherents_type_0.fa", pairedEnd=TRUE )
human_conditions<-c("c1","c1","c2", "c2")
res<-diffExpressedVariants(snp, human_conditions, pvalue=1)
write.table(res$finalTable, file="kissDE_output", sep="\t", quote=FALSE)
Running KisSplice2RefTranscriptome
kissplice2reftranscriptome
-b Trinity.fasta.transdecoder.bed
-k result_coherent_type0.fa
-t out_blat_SNP_on_trinity_ID_80.psl
-s kissDE_output
-o mainOutput.tsv
-l lowQueryCoverageOutput.tsv
The several output files can be found here :
mainOutput.tsv
lowQueryCoverageOutput.tsv