Complementarity of assembly-first and mapping-first approaches for alternative splicing annotation and differential analysis from RNAseq data.
Clara Benoit-Pilven et al.
Support webpage
Abstract: Genome-wide analyses reveal that more than 90% of multi exonic human genes produce at least two transcripts through alternative splicing (AS). Various bioinformatics methods are available to analyze AS from RNAseq data. Most methods start by mapping the reads to an annotated reference genome, but some start by a de novo assembly of the reads. In this paper, we present a systematic comparison of a mapping-first approach (FaRLine) and an assembly-first approach (KisSplice). We applied these methods to two independent RNAseq datasets and found that the predictions of the two pipelines overlapped (70% of exon skipping events were common), but with noticeable differences. The assembly-first approach allowed to find more novel variants, including novel unannotated exons and splice sites. It also predicted AS in recently duplicated genes. The mapping-first approach allowed to find more lowly expressed splicing variants, and splice variants overlapping repeats. This work demonstrates that annotating AS with a single approach leads to missing out a large number of candidates, many of which are differentially regulated across conditions and can be validated experimentally. We therefore advocate for the combine use of both mapping-first and assembly-first approaches for the annotation and differential analysis of AS from RNAseq datasets.
Running and combining the output of KisSplice and FaRLine
I would like information to:
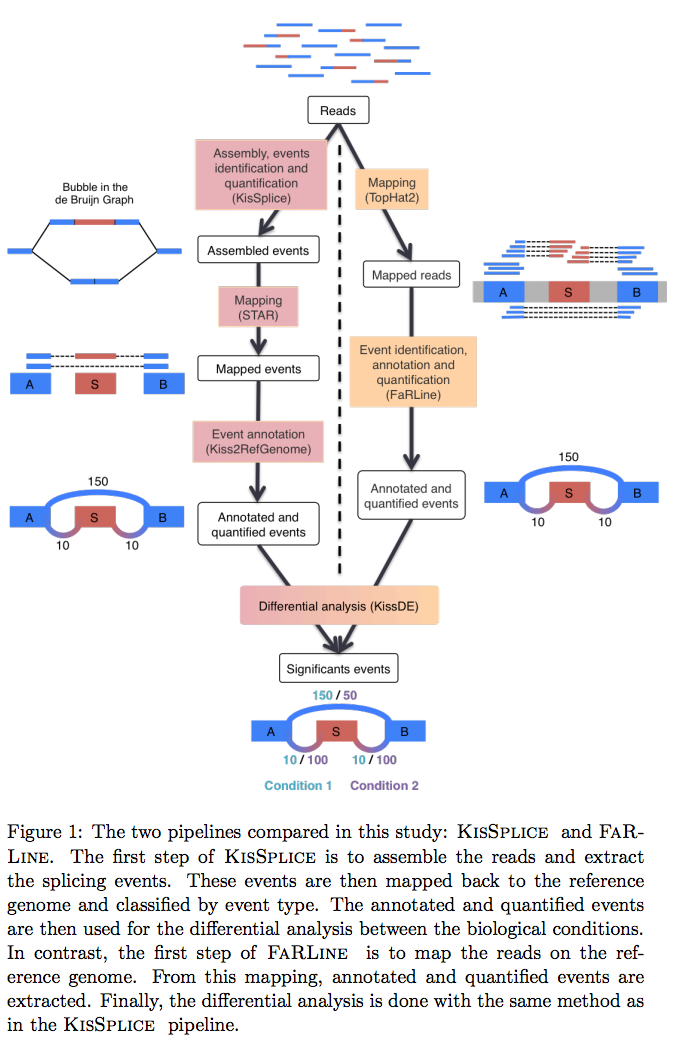
Run KisSplice
Run FaRLine
View interactive venn diagrams comparing KisSplice, FaRLine, MISO, Trinity and Cufflinks
On which dataset?
SKNSH dataset
MCF7 dataset
KisSplice on SKNSH data
The data
We dowloaded a total 959M reads from http://genome.crg.es/encode_RNA_dashboard/hg19/.
They corresponded to long polyA+ RNAs generated by the Gingeras lab, and are also accessible with the following accession numbers (ENCSR000CPN - SRA: SRR315315, SRR315316 and ENCSR000CTT -SRA : SRR534309, SRR534310). For cell lines treated by retinoic acid, the reads were 76nt long, while they were 100nt long for the non treated cells. Hence we trimmed all reads to 76nt.
Running KisSplice
The KisSplice version used in the paper was Version 2.4.0-p1, which can be downloaded here: KisSplice v2.4.0-p1
To replicate the results obtained in the paper, run KisSplice as:
The file containing the bubbles corresponding to alternative splicing events can be found in results/*_coherents_type_1.fa, which from this point on will be referenced as <KisSpliceASFile>. Alternatively, you can download this file here: results_..._coherents_type_1.fa.
To build the STAR index of the genome in a given directory, do:
Alternatively, you can get the index already built here: STAR index
To align the alternative splicing events to the reference genome, do:
The output will be a SAM file SKNSHC0.02_type1_mapped2GRCh37Ensembl75Aligned.out.sam.
Alternatively, you can get the alignment already done here:SKNSHC0.02_type1_mapped2GRCh37Ensembl75Aligned.out.sam
To run kissDE, do:
The output will be a file kissplice_v2.4.0p1_k2rg_kissDE_sknsh.tsv. Alternatively, you can get kissDE output here: kissplice_v2.4.0p1_k2rg_kissDE_sknsh.tsv.
KisSplice on MCF7 data
The data
This data comes from a breast cancer cell line, MCF7. Two conditions were sequenced in duplicated. The cell line was transfected with a siRNA targeting 2 RNA helicase (DDX5 and DDX17) inducing their depletion. The second condition was a control transfection with a siRNA targeting the luciferase firefly gene (GL2). The sequencing was done with the paired-end Illumina technology.
The data can be downloaded here: MCF7 dataset.
Running KisSplice
The KisSplice version used in the paper was Version 2.4.0-p1, which can be downloaded here: KisSplice v2.4.0-p1
To replicate the results obtained in the paper, run KisSplice as:
The file containing the bubbles corresponding to alternative splicing events can be found in results/*_coherents_type_1.fa, which from this point on will be referenced as <KisSpliceASFile>. Alternatively, you can download this file here: results_..._coherents_type_1.fa.
To build the STAR index of the genome in a given directory, do:
Alternatively, you can get the index already built here: STAR index
To align the alternative splicing events to the reference genome, do:
The output will be a SAM file MCF7C0.02_type1_mapped2GRCh37Ensembl75Aligned.out.sam.
Alternatively, you can get the alignment already done here:MCF7C0.02_type1_mapped2GRCh37Ensembl75Aligned.out.sam
To run kissDE, do:
The output will be a file kissplice_v2.4.0p1_k2rg_kissDE_mcf7.tsv. Alternatively, you can get kissDE output here: kissplice_v2.4.0p1_k2rg_kissDE_mcf7.tsv.
FaRLine on SKNSH data
The data
We dowloaded a total 959M reads from http://genome.crg.es/encode_RNA_dashboard/hg19/.
They corresponded to long polyA+ RNAs generated by the Gingeras lab, and are also accessible with the following accession numbers (ENCSR000CPN - SRA: SRR315315, SRR315316 and ENCSR000CTT -SRA : SRR534309, SRR534310). For cell lines treated by retinoic acid, the reads were 76nt long, while they were 100nt long for the non treated cells. Hence we trimmed all reads to 76nt.
Running FaRLine
Downloading
The FaRLine version used in the paper can be downloaded here: FaRLine
We recommend to use FaRLine preferrably on Ubuntu 14.04 LTS or later.
Installing
Extract Farline.tgz in the directory of your choice:
This will create a FARLINE directory containing several files and subdirectories:
FaRLine needs the following dependences to work correctly:
You can easily install all dependencies with the following two scripts:
Your system should now be ready to run Farline properly.
Configuring your run
FaRLine needs a configuration file in order to run. For the SKNSH dataset, you can use as model the file [yourpath]/FARLINE/Farline_SKNSH.conf.
You will need still to set some run and install parameters in the configuration file:
Other parameters are specific for the run and can be ignored.
Launching FaRLine
In order to launch FaRLine, use the following command-line:
you can also add [yourpath]/FARLINE/scripts to your path with the command (or add this line in your .bashrc and source it):
If you do so, the command will be:
To display all available options please use the command:
Launching FaRLine on SKNSH dataset
To replicate the results obtained in the paper, FaRLine can be run in three steps.
Mapping, need fastq files.
Farline computations, need Step1 results.
Stats, need Step2 results
Any part can be run independently if the results from the previous step is available.
Running the 3 steps
To launch all 3 steps, it is essential to define the variable pathfastq in the configuration file, and then run:
Running Step2 and Step3 only
If you wish to skip Step1, you can directly launch Step2 and Step3.
To do so, you need the output of Step1, which can be downloaded here: SKNSH bam files
And to define the variable pathtophat, containing the mapping results from Step1, in the configuration file.
Then run:
Running Step3 only
If you wish to skip Step1 and Step2, you can directly launch Step3.
To do so, you need the output of Step2, which can be downloaded here: SKNSH Step 2 output. Extract the files in this archive.
And to define the variable pathresults in the configuration file to point to the output of Step2.
Then run:
Main output file
After running all the three steps, the main output can be found in the file named exon_skipping_stats_recap_file_SKNSH.xls.
Alternatively, you can download the main output file here: exon_skipping_stats_recap_file_SKNSH.xls.
Troubleshooting
If your ubuntu version is older than 14.04 or if you encounter difficulties to install requested dependencies, you may upgrade Ubuntu with theses commands (use at your own risk, upgrading your Ubuntu version may cause other installed softwares to stop working):
FaRLine on MCF7 data
The data
This data comes from a breast cancer cell line, MCF7. Two conditions were sequenced in duplicated. The cell line was transfected with a siRNA targeting 2 RNA helicase (DDX5 and DDX17) inducing their depletion. The second condition was a control transfection with a siRNA targeting the luciferase firefly gene (GL2). The sequencing was done with the paired-end Illumina technology.
The data can be downloaded here: MCF7 dataset.
Running FaRLine
Downloading
The FaRLine version used in the paper can be downloaded here: FaRLine
We recommend to use FaRLine preferrably on Ubuntu 14.04 LTS or later.
Installing
Extract Farline.tgz in the directory of your choice:
This will create a FARLINE directory containing several files and subdirectories:
FaRLine needs the following dependences to work correctly:
You can easily install all dependencies with the following two scripts:
Your system should now be ready to run Farline properly.
Configuring your run
FaRLine needs a configuration file in order to run. For the MCF7 dataset, you can use as model the file [yourpath]/FARLINE/Farline_MCF7.conf.
You will need still to set some run and install parameters in the configuration file:
Other parameters are specific for the run and can be ignored.
Launching FaRLine
In order to launch FaRLine, use the following command-line:
you can also add [yourpath]/FARLINE/scripts to your path with the command (or add this line in your .bashrc and source it):
If you do so, the command will be:
To display all available options please use the command:
Launching FaRLine on MCF7 dataset
To replicate the results obtained in the paper, FaRLine can be run in three steps.
Mapping, need fastq files.
Farline computations, need Step1 results.
Stats, need Step2 results
Any part can be run independently if the results from the previous step is available.
Running the 3 steps
To launch all 3 steps, it is essential to define the variable pathfastq in the configuration file, and then run:
Running Step2 and Step3 only
If you wish to skip Step1, you can directly launch Step2 and Step3.
To do so, you need the output of Step1, which can be downloaded here: MCF7 bam files.
And to define the variable pathtophat, containing the mapping results from Step1, in the configuration file.
Then run:
Running Step3 only
If you wish to skip Step1 and Step2, you can directly launch Step3.
To do so, you need the output of Step2, which can be downloaded here: MCF7 Step 2 output. Extract the files in this archive.
And to define the variable pathresults in the configuration file to point to the output of Step2.
Then run:
Main output file
After running all the three steps, the main output can be found in the file named exon_skipping_stats_recap_file_MCF7.xls.
Alternatively, you can download the main output file here: exon_skipping_stats_recap_file_MCF7.xls.
Troubleshooting
If your ubuntu version is older than 14.04 or if you encounter difficulties to install requested dependencies, you may upgrade Ubuntu with theses commands (use at your own risk, upgrading your Ubuntu version may cause other installed softwares to stop working):
Select the human genome hg19 (from the first drop-down menu)
Load the sorted bam files: File > Load from File... . Select all the sorted bam files you want to load.
Use Ctrl in Windows or Linux and Command in Mac to select several bam files at once.
These bam files must have a .bai file associated (created in the Index step).
Analyse an event by typing its Gene name, or its genomic position, and pressing Go.
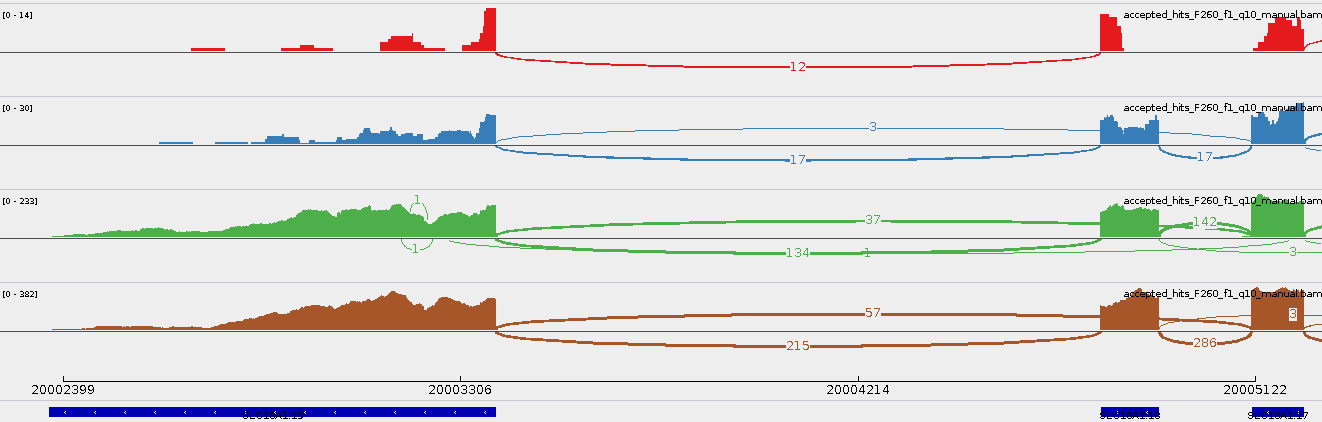
A handy way of doing this is zooming into your event of interest and viewing the Sashimi Plot (right-click on one the bam track (left panel), and select Sashimi Plot).
As an example, you can view here the Sashimi Plot of SLC18A1 18 17:19 (the first event found by all methods in the SKNSH dataset).
For an example of events with novel exons, here is the Sashimi Plot of ZNF236 2 3:i1, where the flanking non-annotated exon is in intron 1 (see the black box).
Interactive Venn diagram of the Exon Skipping events found by the five methods on the SKNSH dataset:
*The annotations considered for these events (gene name, exon coordinates, exon numbering, etc) can be either visualised here: FasterDB - EnsEMBL r75 Browser or can be downloaded as a BED file here: FasterDB annotations
No methods selected
Scroll down for tips on how to use this Venn diagram and on how to analyse an event using IGV/Sashimi plots.
Tips:
Hovering the cursor on the numbers will highlight its corresponding methods;
Clicking on the numbers will give you the list of Exon Skipping events found by the corresponding methods;
The Sashimi Plot of an event can help you to understand it better. For example, in the following you see can the Sashimi Plot of SLC18A1 18 17:19 (the first event found by all methods in the SKNSH dataset).
Interactive Venn diagram of the Exon Skipping events found by the five methods on the MCF7 dataset:
*The annotations considered for these events (gene name, exon coordinates, exon numbering, etc) can be either visualised here: FasterDB - EnsEMBL r75 Browser or can be downloaded as a BED file here: FasterDB annotations
No methods selected
Scroll down for tips on how to use this Venn diagram and on how to analyse an event using IGV/Sashimi plots.
Tips:
Hovering the cursor on the numbers will highlight its corresponding methods;
Clicking on the numbers will give you the list of Exon Skipping events found by the corresponding methods;
The Sashimi Plot of an event can help you to understand it better. For example, in the following you see can the Sashimi Plot of SLC18A1 18 17:19 (the first event found by all methods in the SKNSH dataset).
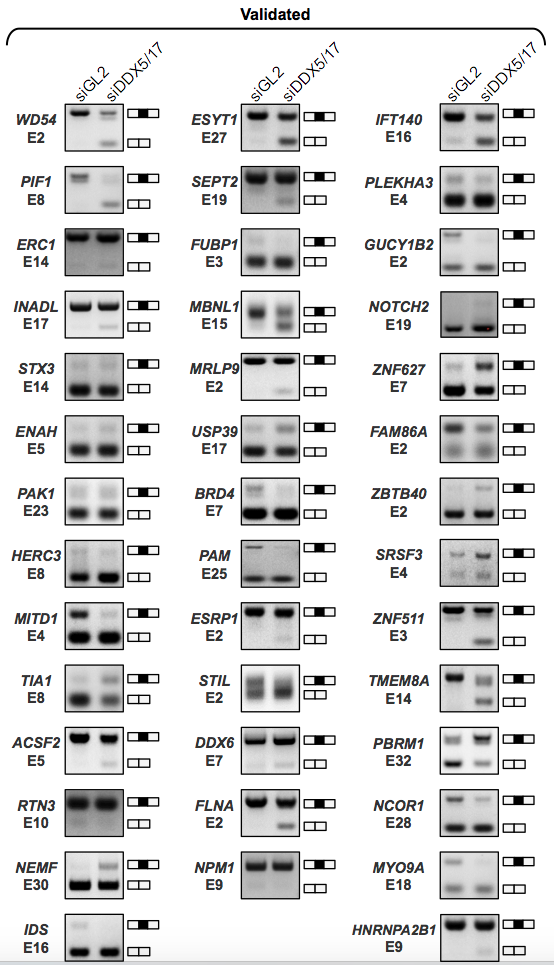
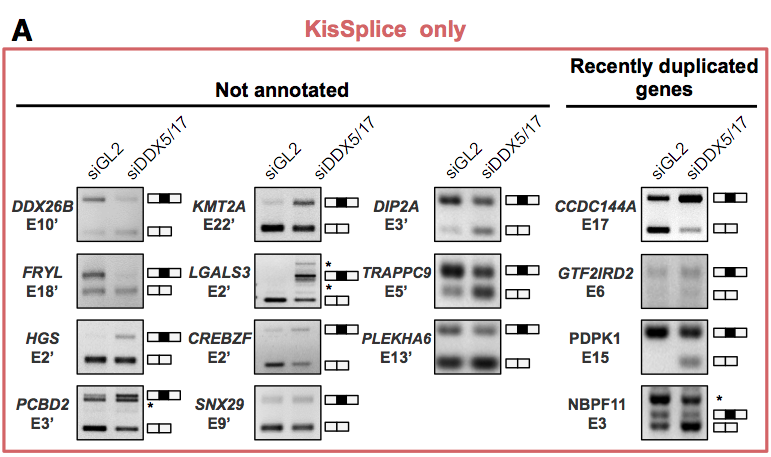
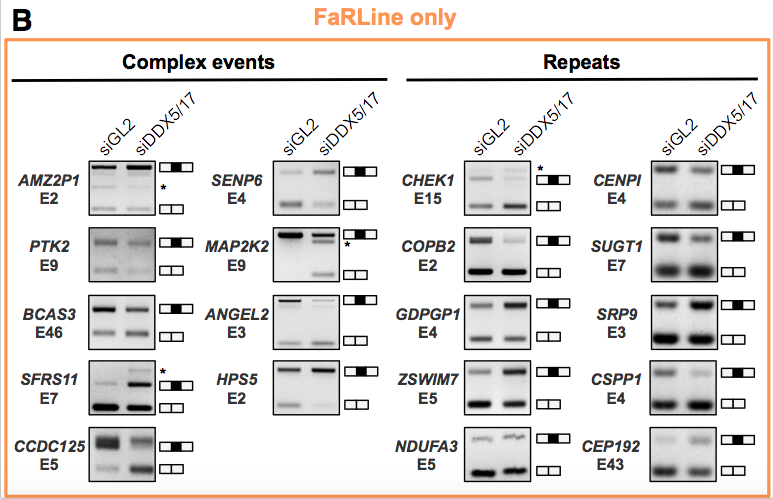
RT-PCR validations of events (only from KisSplice and FaRLine on the MCF7 dataset) Common events:
 This step takes around 30 GB of RAM.
This step takes around 30 GB of RAM.